
![]()
Luca Gallelli – Prof. Aggregato di Farmacologia, Scuola di Medicina, Università di Catanzaro
La scadenza dei brevetti che coprono molti farmaci di riconosciuta efficacia e sicurezza ha aperto la strada alla produzione di “copie” che vengono immesse sul mercato generalmente a prezzi più bassi rispetto agli originali (“originators”). Si tratta dei cosiddetti farmaci “ generici” o, secondo la dizione più appropriata, “bioequivalenti”. Per bioequivalente si intende, propriamente: “Un medicinale che ha la stessa composizione qualitativa e quantitativa di sostanze attive e la stessa forma farmaceutica del medicinale di riferimento, nonché una bioequivalenza con il medicinale di riferimento, dimostrata da studi appropriati di biodisponibilità” (1).
Come si può notare, tale definizione pone l’accento sull’importanza determinante dell’esistenza di appropriati studi di bioequivalenza perché un farmaco “copia” possa essere ritenuto equivalente all’“originator”. Il parametro che viene studiato è essenzialmente la biodisponibilità, che definisce la percentuale della dose somministrata che effettivamente entra nella circolazione sistemica. In pratica, gli studi di bioequivalenza hanno la finalità di dimostrare che le differenze di biodisponibilità del principio attivo, che inevitabilmente esistono tra l’“originator” e la sua “copia”, non superino un certo intervallo di variazione.
Gli studi di bioequivalenza possono essere eseguiti sia mediante la somministrazione di una singola dose (crossover) che di multiple dosi successive. Essi in genere vengono condotti su un campione piuttosto limitato di volontari sani (minimo 12 soggetti di età compresa tra i 18 ed i 55 anni) di ambedue i sessi e non utilizzano parametri clinici di efficacia, bensì si limitano a confrontare la biodisponibilità farmacologica sistemica di due prodotti, tramite il confronto statistico di alcuni parametri farmacocinetici (2), tra cui:
- Area sotto la curva concentrazione/tempo (AUC);
- Concentrazione massima raggiunta nel plasma (Cmax),
- Tempo in cui viene raggiunta questa concentrazione (tmax).
Quando si mettono a confronto due prodotti con lo stesso principio attivo, esse sono definite bioequivalenti se la differenza tra le rispettive biodisponibilità rientra in un certo intervallo, predefinito come “intervallo accettabile” e convenzionalmente fissato nel range ± 20% .
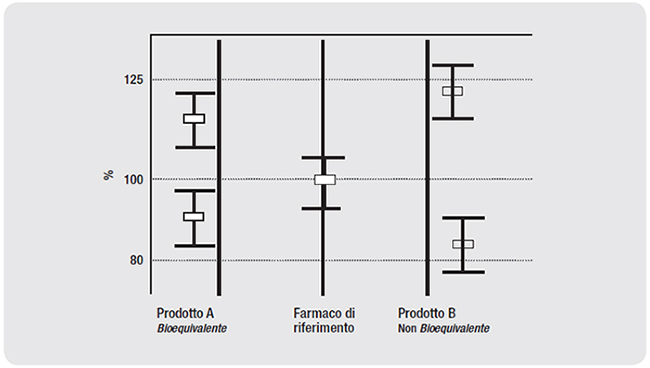
Più precisamente, in termini statistici, un farmaco è considerato bioequivalente se l’“intervallo di confidenza al 90%” della sua biodisponibilità rientra nell’80%-125% del farmaco di riferimento (3) (Figura 1).
Figura 1
Negli studi di bioequivalenza, un prodotto è definito come bioequivalente al farmaco di riferimento, se il suo intervallo di confidenza al 90% rientra nell’80%-125% del farmaco di riferimento.
Va ricordato però che esistono limiti insiti nella natura stessa degli studi di bioequivalenza e che possono ripercuotersi sull’affidabilità del prodotto nella pratica clinica. In primo luogo, convenzionalmente si ritiene che l’intervallo “accettabile” di bioequivalenza corrisponda ad una analoga “accettabilità” dell’equivalenza terapeutica; in realtà, si tratta di un intervallo piuttosto ampio e, soprattutto, non differenziato per categoria terapeutica e classe farmacologica, che tende a trascurare le diverse variabili farmacologiche e cliniche che possono incidere significativamente sull’equivalenza terapeutica di due prodotti. Il problema della non costante corrispondenza tra “equivalenza farmacocinetica” (bioequivalenza) ed “equivalenza terapeutica” può essere particolarmente rilevante per i farmaci che hanno una “finestra terapeutica” piuttosto ristretta.
In questi casi, malgrado la differenza di biodisponibilità sia fissata nel range ± 10%, (in America è ± 5%) passare da un prodotto all’altro, pur a parità teorica di principio attivo, potrebbe correlarsi più facilmente con l’inefficacia terapeutica o con lo sviluppo di eventi avversi. Inoltre, il numero piuttosto limitato di soggetti che viene arruolato negli studi di bioequivalenza non consente di stabilire con assoluta certezza che la variabilità dell’intervallo di biodisponibilità non possa essere, in una popolazione più vasta, ben più ampio di quello che viene evidenziato in studi di piccole dimensioni.
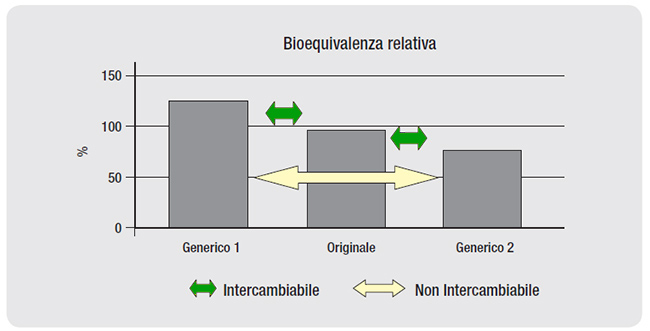
Esiste inoltre un’altra problematica, che riguarda la sostituibilità tra due diversi farmaci generici dello stesso “originator”, nota come “fenomeno bio-creep”.
Esso deriva dal fatto che i test di bioequivalenza confrontano ciascun generico con il suo corrispondente prodotto originale, ma non confrontano tra i loro diversi generici. Pertanto, la sostituibilità potrebbe avvenire teoricamente solo tra farmaco originale e farmaco generico, ma non tra due formulazioni di generici; infatti il concetto di bioequivalenza non gode della “proprietà transitiva”, cioè non è esatto ritenere che due prodotti, ciascuno bioequivalente con lo stesso standard di riferimento, siano bioequivalenti tra di loro, se non viene effettuato un confronto diretto (cosa che non avviene) (4) (Figura 2).
Inoltre, non si fa riferimento al ruolo degli eccipienti che non solo svolgono un ruolo importante nei processi di assorbimento e distribuzione del farmaco, quanto possono essere responsabili dello sviluppo di eventi avversi (basti pensare a ciò che potrebbe indurre il lattosio se assunto da pazienti intolleranti).
Di conseguenza, il passaggio da un farmaco equivalente ad un altro può associarsi ad importanti rischi per il paziente, anche in termini di tollerabilità (5). Pertanto lo switch tra farmaci equivalenti non dovrebbe essere un processo automatico ma dovrebbe essere ben valutato dal medico.
Figura 2
Fenomeno del “bio-creep”. Entrambi i generici sono intercambiabili con il prodotto originale, mentre i due generici non sono intercambiabili tra di loro.
Bibliografia
1. Direttiva europea 2001/83/CE e successive modificazioni, art. 10, comma 5 DLvo n. 219/06; art. 10, comma 2
2. Hardman JH, Limbird LE (Editors). Goodman&Gilman’s -The Pharmacological Basis of Therapeutics, Tenth Edition. McGrawHill 2001
3. Meredith P. Bioequivalence and other unresolved issues in generic drug substitution. Clin Ther. 2003 Nov; 25(11):2875-90
4. Dong-Seok Yim. Simulation of the AUC changes after generic substitution in patients. J Korean Med Sci 2009; 24:7-12
5. Dueñas-Laita A, Pineda F, Armentia A. Hypersensitivity to generic
drugs with soybean oil. N Engl J Med. 2009 Sep 24; 361(13):1317-8
Sommario - Qualche termine di farmacocinetica
|
Qualche termine di farmacocinetica... La farmacocinetica è la branca della farmacologia che studia in termini quantitativi l’assorbimento, la distribuzione, il metabolismo e l’eliminazione dei farmaci. Di seguito, forniamo la definizione di alcuni parametri farmacocinetici generalmente utilizzati negli studi di bioequivalenza (Figura 1) AUC: parametro farmacocinetico che indica l’area sotto la curva della concentrazione plasmatica di un farmaco rispetto al tempo. Biodisponibilità: questo termine viene utilizzato per descrivere sia la frazione di un farmaco somministrato che raggiunge la circolazione sistemica senza subire alcuna modificazione chimica rispetto al totale di farmaco somministrato, sia la velocità con cui un farmaco è reso disponibile nella circolazione sistemica.... Leggi altro |
|

N.11/2013 - MedTOPICS - Periodico Quindicinale
È vietata la riproduzione totale o parziale senza il consenso scritto dell'editore. - 13T0375
Con il contributo non condizionante di A. Menarini IFR
Copyright © 2013
Colophon | Informazioni legali