
LA SIMILIRITA' FARMACOCINETICA NELLA SOSTITUIBILITA' DEGLI ANTIEPILETTICI
Abstract
There is considerable interest and debate concerning the place of generic substitution, especially relating to antiepileptic drugs (AEDs). A careful review of the literature revealed no adequately powered randomized controlled trials that assessed the risk/benefit ratio of generic substitution. The conflicting results create confusion, often intentionally fueled by the pharmaceutical industry of branded products among health professionals and patients regarding the regulatory determination of bioequivalence. As stated by the Italian League against Epilepsy (LICE), generic AEDs meeting current regulatory criteria for bioequivalence represent a valuable choice in the management of epilepsy by allowing a substantial reduction of treatment costs, particularly in patients initiating monotherapy or adjunctive treament and in those with persistent seizures. Reportedly, in patients who achieved seizure freedom a modest change in plasma drug levels, which may occasionally occur even after substitution of products that meet bioequivalence criteria, could in rare cases lead to seizure breakthrough. Additional criteria should be introduced for making the best therapeutic decision when different generic AEDs are commercially available. One of these is the concept of pharmacokinetic similarity, the topic of the present article.
Introduzione
Negli ultimi anni un considerevole interesse ha destato, non solo nella comunità scientifica, ma anche nell’opinione pubblica, il dibattito sulle basi farmacologiche e cliniche della sostituibilità del farmaco a brevetto scaduto (1,2). Le cause della netta contrapposizione tra i sostenitori e i detrattori della sostituibilità sono la scarsa conoscenza dell’argomento, spesso alimentata dalle aziende farmaceutiche del prodotto di marca, che ambiscono a preservare il monopolio del marcato nonostante l’ingresso commerciale dei corrispondenti generici, che hanno il vantaggio di costare meno (3). La bioequivalenza, il principale criterio che garantisce che un prodotto generico abbia lo stesso profilo di efficacia e sicurezza del prodotto originatore (cioè la stessa equivalenza terapeutica), non è solo una nozione di chimica farmaceutica, ma anche un concetto di farmacologia clinica con implicazioni medico-cliniche (4-6). In generale il diffuso scetticismo dei medici prescrittori, nei confronti dei farmaci generici (o, con terminologia meno stigmatizzante, equivalenti), deriva essenzialmente da considerazioni aneddotiche e studi clinici di scarsa validità scientifica, spesso sponsorizzati, che suggerirebbero l’inaffidabilità dei farmaci equivalenti in termini di efficacia e sicurezza (3). Il problema della sostituibilità (e quindi interscambiabilità) nei pazienti epilettici che abbiano ottenuto la remissione clinica completa, seppur ampiamente dibattuto in letteratura biomedica, rimane tuttora irrisolto e richiederà la conduzione di studi clinici controllati e randomizzati che stabiliscano, con adeguata potenza statistica, il rapporto rischio/beneficio del prodotto equivalente con quello originatore. Tuttavia, come dichiarato da molte organizzazioni scientifiche di rilevanza nazionale e internazionale, tra cui la Lega Italiana contro l’Epilessia (LICE), numerose sono le circostanze in cui l’antiepilettico equivalente potrebbe essere prescritto con lo stesso rapporto di rischio/efficacia del relativo farmaco originatore: scelta del medicinale (originatore o generico) all’inizio della terapia antiepilettica nel paziente naïve, terapia di combinazione nel paziente epilettico resistente o insufficientemente controllato dalla monoterapia, sostituzione della monoterapia del paziente epilettico resistente ecc (7). Sebbene gli antiepilettici non siano farmaci particolarmente costosi rispetto ad altri, a causa dell’elevata prevalenza e della malattia e la lunga durata del trattamento, la spesa farmaceutica relativa all’epilessia non è trascurabile (8-11). Negli ultimi anni la crescita esponenziale della spesa sanitaria, riconducibile all’aumento della popolazione geriatrica e all’introduzione di nuove terapie particolarmente costose (per es., i farmaci biologici in ambito oncologico), ha costretto i governi dei principali Paesi occidentali ad adottare urgenti misure per ridurre i costi. Una delle più efficaci strategie per ridurre i costi diretti si è rivelata la promozione alla prescrizione “razionale” del farmaco generico. È stato dimostrato che la prescrizione dei generici è associata a un consistente risparmio per le casse del Sistema Sanitario Nazionale (SSN) e dei cittadini. Al riguardo, in Italia, in cui la prescrizione del generico è estremamente limitata rispetto agli altri Paesi europei, l’introduzione dei farmaci generici ha consentito di risparmiare almeno 25 milioni di euro nel 2002 (12). Si stima che il risparmio derivante da un uso più diffuso della sostituibilità da parte dei medici prescrittori potrebbe raggiungere l’11% della spesa farmaceutica annuale negli USA (9). Diversi studi hanno evidenziato come un aumento del co-payment dei farmaci (cioè la differenza dei costi per la singola confezione tra il farmaco originatore e il corrispondente generico, che è a carico del paziente) si associ ad una riduzione della prescrizione e dell’aderenza terapeutica anche per farmaci molto efficaci impiegati nella gestione di condizioni croniche (13-16). Pertanto, la politica sanitaria (ma anche il singolo medico) dovrebbe ridurre il ricorso al co-payment, aumentando l’aderenza alle terapie. Nel presente articolo sarà richiamato il concetto di bioequivalenza, evidenziando i limiti della relazione tra bioequivalenza ed equivalenza terapeutica e quindi la necessità di considerare altri parametri di valutazione, tra cui la similarità farmacocinetica, nella scelta (razionale) del farmaco equivalente e, più specificamente, dell’antiepilettico (12). Il concetto di similarità farmacocinetica potrebbe aiutare il medico prescrittore non solo a scegliere l’antiepilettico più appropriato, ma, per il minor costo della terapia, a massimizzare l’aderenza terapeutica e ridurre la spesa sanitaria, con innegabili vantaggi per il singolo paziente e la collettività.
Quando un prodotto si definisce equivalente?
Il produttore di un medicinale equivalente, per ottenerne l’autorizzazione all’immissione in commercio, è dispensato, salvo casi particolari, dal presentare studi di efficacia e sicurezza in quanto la molecola è già nota. Secondo l’attuale normativa vigente occorre produrre dati che dimostrino l’equivalenza farmaceutica e, nel caso che la formulazione in domanda preveda un processo di assorbimento dal sito di somministrazione, la bioequivalenza con il medicinale di riferimento (originatore). Non vi è assolutamente ragione scientifica per ipotizzare diversa efficacia o sicurezza (cioè differente equivalenza terapeutica), se il generico viene prodotto nel rispetto dei principi (scientifici e normativi) dell’equivalenza farmaceutica e della bioequivalenza (17). Due prodotti che contengono lo stesso principio attivo in termini qualitativi (molecola) e quantitativi (dose) e che, come preparazione farmaceutica, sono destinati alla stessa via di somministrazione si definiscono equivalenti farmaceutici. Un prodotto generico deve rispettare rigidi standard di identità, qualità e purezza per essere definito un equivalente farmaceutico; tuttavia, un generico può differire dall’originatore in molte caratteristiche: forma, meccanismo di rilascio, impacchettamento, eccipienti (tra cui coloranti, conservanti, edulcoranti ecc), data di scadenza e, entro certi limiti, etichettatura. È importante precisare che gli equivalenti farmaceutici non sono necessariamente bioequivalenti (18). Per dimostrare la bioequivalenza di un prodotto, si ricorre allo studio farmacocinetico di bioequivalenza (in vivo). Il quesito scientifico non riguarda l’efficacia della molecola, che è già nota, ma la performance farmaceutica e quindi il profilo farmacocinetico della specifica formulazione del farmaco equivalente. Precisamente, due equivalenti farmaceutici le cui velocità ed entità di assorbimento non differiscono statisticamente, quando essi sono somministrati alla stessa dose e in condizioni sperimentali simili, sono definiti bioequivalenti (Figura 1) (17,18). Per dimostrare la bioequivalenza, si confronta il valore medio di alcuni parametri farmacocinetici ottenuti dalla misurazione delle concentrazioni ematiche del farmaco in un gruppo di volontari sani (24-36 soggetti), ai quali viene somministrata in due periodi successivi (studio cross-over) una singola dose a digiuno dei due prodotti a confronto (Figura 1). Non esistendo alcun metodo statistico per dimostrare l’uguaglianza di due prodotti, gli studi di bioequivalenza si propongono di verificare l’assenza di una differenza clinicamente rilevante attraverso la stima di una differenza minima ammissibile (cioè “essential similarity”) (18,19).
FIGURA 1. Analisi della bioequivalenza: un ipotetico studio di bioequivalenza che confronta due preparazioni farmaceutiche dello stesso farmaco. |
I parametri farmacocinetici che vengono studiati sono in particolare l’area sotto la curva delle concentrazioni plasmatiche (AUC) e la concentrazione di picco (Cmax). I due parametri devono essere sovrapponibili per entrambi i prodotti, generico e originatore, con un margine di tolleranza del 20%. Due formulazioni sono definite bioequivalenti se l’intervallo di confidenza al 90% del rapporto tra ciascun parametro (AUCgenerico/AUCoriginatore o Cmaxgenerico/Cmaxoriginatore) sia compreso all’interno dei limiti stabiliti dalla normativa, cioè 0.80 e 1.25 (intervallo di bioequivalenza). Questo intervallo è convenzionalmente (e anche arbitrariamente) ritenuto compatibile con l’equivalenza terapeutica. Se i limiti di confidenza cadono al di fuori dall’intervallo prestabilito, il prodotto non viene considerato bioequivalente (Figura 1). In alcuni casi, viene considerato anche il tempo (Tmax) in cui viene raggiunta Cmax (18,19). Teoricamente la variazione fra un prodotto e l’altro potrebbe raggiungere il 45%; in realtà, l’obbligo di presentare un intervallo di confidenza entro i limiti stabiliti spinge i produttori a mantenersi abbastanza vicini al 100%. Infatti, le differenze fra prodotto originatore e generico sono di solito contenute entro il 10% di variazione e più spesso entro il 3% (19).
Limiti della definizione di bioequivalenza
Il problema della sostituibilità farmaco originatore e farmaco equivalente oppure farmaco equivalente e farmaco equivalente è legato strettamente ai limiti della definizione di bioequivalenza (20,21). Gli studi di bioequivalenza non utilizzano parametri clinici di efficacia, bensì si limitano a confrontare la biodisponibilità farmacologica sistemica di due prodotti (cioè AUC e Cmax). Anche se la procedura di determinazione della bioequivalenza è molto rigorosa, rimangono problemi di difficile soluzione (19). Innanzitutto, l’intervallo di bioequivalenza è uno standard stabilito convenzionalmente attribuendo maggior rilievo alla variabilità del comportamento in vivo della formulazione piuttosto che alla variabilità della risposta terapeutica nella popolazione dei pazienti. Un intervallo di bioequivalenza così ampio, non differenziato per categoria terapeutica e per classe farmacologica, tende a trascurare le altre variabili farmacologiche e cliniche che possono incidere significativamente sull’equivalenza terapeutica di due prodotti. Per alcuni prodotti l’intervallo potrebbe essere troppo largo, mentre per altri troppo stretto (22,23). Un altro limite è che la procedura matematico-statistica adottata negli studi di bioequivalenza consente di stimare una bioequivalenza “media”, calcolata su una popolazione. Al medico prescrittore interessa disporre di una bioequivalenza “individuale”, che si riferisce al paziente al quale somministrare il farmaco. Oltre al valore medio, si dovrebbe considerare la distribuzione dei parametri di biodisponibilità (24). La bioequivalenza “media” coinciderebbe con la bioequivalenza “individuale” se gli indici di distribuzione di AUC e Cmax per i due prodotti a confronto fossero uguali. In caso contrario, le due formulazioni, anche se bioequivalenti dal punto di vista statistico, potrebbero non essere terapeuticamente equivalenti per il singolo paziente a causa della diversità di distribuzione delle biodisponibilità. Seppur difficilmente praticabile, occorre stimare la bioequivalenza individuale, ossia entro soggetto (anche in diverse condizioni), e valutare in quale percentuale i singoli soggetti rispondono (equivalentemente) a entrambi i prodotti, generico e originatore. La biodisponibilità individuale si configura, quindi, come il criterio fondamentale per poter applicare la norma della sostituibilità tra formulazioni nel corso di un trattamento in atto, senza pregiudicare il profilo terapeutico e la sicurezza (25-29). Poiché gli studi di bioequivalenza sono condotti con la somministrazione di una singola dose in soggetti adulti sani volontari, per definizione sono esclusi i pazienti affetti dalla patologia di interesse (epilessia), che, nella realtà clinica, possono presentare comorbidità o essere in trattamento con altri farmaci, che possono introdurre variabilità nel profilo farmacocinetico del prodotto equivalente, contenente differenti eccipienti rispetto al prodotto originatore. Sebbene il disegno dello studio di bioequivalenza sia statisticamente valido, sorge il dubbio se questo approccio normativamente regolamentato sia anche clinicamente appropriato (28). In particolare, si ritiene ragionevole domandarsi se i dati generati dagli studi di bioequivalenza non possano essere generalizzati a tutti i gruppi di pazienti, come per esempio la popolazione geriatrica in cui, come è noto, si osserva un’alterazione di molti parametri farmacocinetici (29).
FIGURA 2. La definizione (normativa) di bioequivalenza non gode della proprietà transitiva. Pertanto, due farmaci generici (per es., 1 e 2) sono equivalenti all’originatore senza essere tra loro equivalenti. Il fenomeno prende il nome di bio-creep. BA = biodisponibilità.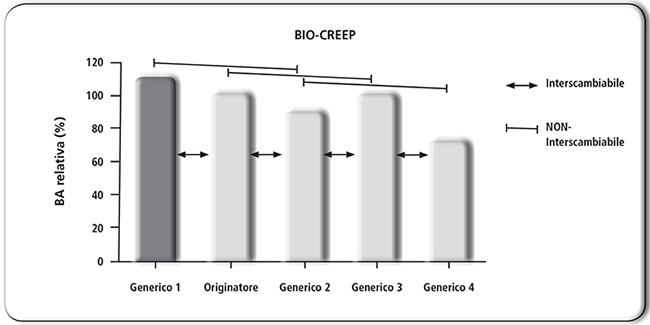 |
L’estrapolazione dei risultati di uno studio di bioequivalenza in singola dose non predice necessariamente il profilo farmacocinetico del farmaco che, somministrato cronicamente, ha raggiunto lo steady-state. Un prodotto equivalente (farmacocineticamente poco performante perché si discosta dal prodotto originatore) può rientrare nell’ (ampio) intervallo di bioequivalenza se somministrato in singola dose, ma non in dosi multiple (30). Un altro problema che può limitare la sostituibilità, soprattutto tra farmaco equivalente e farmaco equivalente dello stesso prodotto originatore, è il fenomeno del cosiddetto “bio-creep” (Figura 2). Infatti, gli studi di bioequivalenza sono condotti confrontando il singolo prodotto equivalente e il prodotto originatore. Questa situazione non garantisce che due o più equivalenti dello stesso originatore siano tra loro bioequivalenti. Per esempio, supponendo che un generico abbia una biodisponibilità (valutata in AUC) +16% ed un secondo generico -14%, entrambi sono bioequivalenti rispetto allo standard con cui sono stati confrontati (originatore), ma non sono tra loro bioequivalenti essendo la differenza tra loro superiore al 20% (31). Da quanto discusso, è facile comprendere che il concetto di bioequivalenza non gode della proprietà transitiva: non è possibile concludere, senza una verifica diretta, che due prodotti, ciascuno bioequivalente con lo stesso standard di riferimento (originatore), siano bioequivalenti tra di loro. Semplicisticamente parlando, se il farmaco 2 è equivalente al farmaco 3 e se il farmaco 4 è equivalente al farmaco 3, non è detto che il farmaco 2 sia equivalente al farmaco 4 (Figura 2) (32). Purtroppo, il confronto diretto non è possibile in quanto i medicinali equivalenti sono (su base normativa!) confrontati unicamente con il medicinale originatore e così l’interscambiabilità (sostituibilità) fra equivalenti è solo supposta.
Differenze farmacocinetiche tra diversi Levetiracetam
La maggior parte dei farmaci antiepilettici è caratterizzata da un ristretto indice terapeutico, cioè la dose terapeutica è spesso vicina alla dose che causa tossicità. È plausibile che riduzioni modeste dei livelli plasmatici, ad esempio, dell’ordine del 20%, possano essere sufficienti a determinare la ricomparsa di una crisi epilettica in pazienti precedentemente controllati dalla terapia (33). Una simile situazione può presentarsi dopo sostituzione di un prodotto farmaceutico con un altro, anche se bioequivalente (34). Questo principio è riconosciuto a livello normativo in alcuni Paesi Europei, come la Germania, che non permettono, per farmaci a ridotto indice terapeutico, la sostituibilità “automatica” con generici da parte del farmacista. Il problema non è limitato solo alla sostituibilità farmaco generico e farmaco originatore (per l’intervallo di bioequivalenza 80-125%), ma anche farmaco generico e farmaco generico (per il fenomeno del bio-creep). Al riguardo, è interessante ricordare che, a differenza di altri agenzie regolatorie del farmaco come EMA e FDA, la Health Canada ha adottato un intervallo di bioequivalenza più stringente (cioè 90-112%) per i farmaci a dose critica, tra cui gli antiepilettici come la fenitoina, caratterizzata da una cinetica di ordine zero (35). In un recente studio, condotto analizzando i dati farmacocinetici inclusi nelle domande di autorizzazione all’immissione in commercio per levetiracetam, presentate da alcuni produttori all’EMA, che pubblica le decisioni adottate dal CHMP (Committee for Medicinal Products for Human Use) dopo le procedure di valutazione, è emersa l’esistenza di una marcata eterogeneità dei singoli prodotti equivalenti rispetto al Keppra®, che rappresenta il prodotto originatore con cui confrontarsi. In particolare, a parità di intervallo di confidenza dei prodotti equivalenti analizzati, Matever® è quello che farmacocineticamente è più simile a Keppra® (con un rapporto di Cmax Matever®/Keppra® ~ 100%) (Figure 3 e 4).
FIGURA 3. A parità di intervallo di confidenza il valore medio del rapporto di Cmax (Cmaxgenerico/Cmaxoriginato) di alcuni equivalenti di levetiracetam, commercialmente disponibili, appare estremamente differente. Nell’analisi comparativa sono stati inclusi solo gli equivalenti di levetiracetam la cui autorizzazione all’immissione commerciale è stata concessa con procedura centralizzata nel rispetto della normativa EMA in tema di bioequivalenza, con pubblicazione dei parametri farmacocinetici.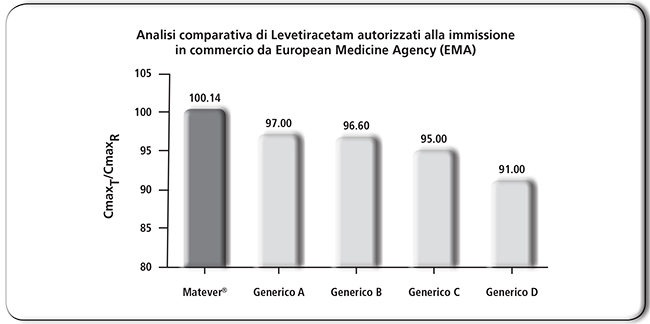 |
FIGURA 4. Un prodotto contenente levetiracetam si definisce equivalente se i parametri farmacocinetici rientrano nell’intervallo di bioequivalenza. Tuttavia, alcuni equivalenti di levetiracetam, tra cui Matever®, hanno il vantaggio di possedere una più spiccata similarità farmacocinetica: a parità di intervallo di confidenza, la media coincide con il 100% (rispetto all’originatore, cioè Keppra®).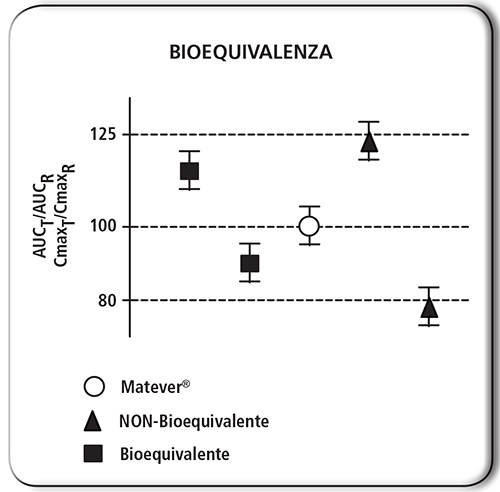 |
È importante notare che il concetto di bioequivalenza è di tipo normativo e, quindi, un prodotto (come tutti i generici di levetiracetam attualmente disponibili in commercio), se rispetta l’intervallo di bioequivalenza, si definisce (ex-lege) equivalente. Pertanto, non si può parlare di prodotto più o meno equivalente, ma solo di prodotto equivalente o non equivalente. Tuttavia, il concetto di “similarità farmacocinetica” può costituire un criterio aggiuntivo che il medico prescrittore o il farmacista dispensatore possono adottare nella selezione del prodotto all’atto della sostituzione (31). La pubblicazione dei parametri farmacocinetici e dei rapporti di bioequivalenza dei prodotti generici nella scheda tecnica dovrebbe essere resa normativamente obbligatoria. Qualcuno potrebbe obiettare che i limiti dell’intervallo di bioequivalenza sono già restrittivi se si considerano le variazioni inter- o intra-individuali delle concentrazioni plasmatiche del farmaco o i risultati delle prove di qualità dei lotti dello stesso prodotto. Tuttavia, questo può non essere sufficiente per i farmaci a ristretto indice terapeutico o appartenenti a particolari categorie farmacologiche come gli antiepilettici (22). Al riguardo, a causa delle implicazioni psicologiche, mediche (trauma cranico per caduta), sociali (alcune attività lavorative rischiose) e normative (patente di guida) della ricomparsa di crisi epilettiche, precedentemente controllate, è auspicabile che sia attuata ogni misura ragionevole per minimizzare il rischio di recidiva nei pazienti che abbiano ottenuto la remissione clinica completa. Attualmente, in assenza di studi controllati, randomizzati e in doppio cieco, che dimostrino la sostituibilità dell’antiepilettico originatore (Keppra®), la similarità farmacocinetica è un valore aggiunto che non può essere trascurato nell’atto decisionale del più appropriato prodotto per il singolo paziente affetto da epilessia (7).
Conclusioni
Il dibattito della sostituibilità dell’antiepilettico originatore vs. generico o generico vs. generico continuerà finché studi clinici con appropriato disegno sperimentale e adeguata potenza statistica definiranno il rapporto rischio/beneficio dell’interscambiabilità del prodotto antiepilettico (originatore e generico) per il paziente, che include la valutazione non solo della frequenza di crisi epilettiche dopo remissione completa, ma anche del profilo di sicurezza. Solo con l’approccio della medicina basata sull’evidenza sarà possibile superare i limiti della definizione (normativa) di bioequivalenza, tra cui l’equivalenza terapeutica di popolazione vs. del singolo individuo oppure il fenomeno del bio-creep (36). In attesa di risultati scientificamente validati, la proposta di considerare la similarità farmacocinetica come valore aggiunto per differenziare e quindi selezionare l’antiepilettico equivalente più confacente alle necessità clinico-terapeutiche del singolo paziente può essere accolta favorevolmente anche dai medici e pazienti più scettici, che intendono beneficiare del (consistente) risparmio economico associato all’acquisto del generico. L’esempio di Matever® dimostra l’esistenza di un’eterogeneità di performance farmaceutica e quindi farmacocinetica tra i numerosi equivalenti di levetiracetam, disponibili commercialmente, che potrebbe (forse) giustificare l’adozione di una sostituibilità “regolamentata” anche sulla base della similarità farmacocinetica.
|