
Biosimilari: qualità e sostenibilità, il caso dell’epoetina zeta
Quando si parla di farmaci biotecnologici e biosimilari, la prima distinzione da fare è di ordine terminologico.
Per “farmaci biotecnologici” si intendono, in generale, medicinali contenenti uno o più principi attivi prodotti o ricavati da organismi viventi, rappresentati principalmente da cellule procariotiche o eucariotiche, opportunamente modificate mediante la tecnologia dell’ingegneria genetica.
Si tratta di molecole complesse di grandi dimensioni, costituite da miscele eterogenee di biomolecole diverse tra loro dal punto di vista del peso molecolare (p.m.) e che presentano variazioni legate sia alla naturale biodiversità del processo naturale di sintesi da parte di organismi viventi, sia allo stesso processo produttivo industriale.
Nel contesto dei farmaci biotecnologici, risulta in effetti assolutamente giustificato l’aforisma secondo cui “the process is the product” (“il processo è il prodotto”).
La produzione di un farmaco biotecnologico è un processo complesso, che avviene attraverso una ben articolata sequenza di fasi, che includono:
• la definizione e lo sviluppo di una cellula ospite;
• la creazione di una banca cellulare (Working Cell Bank);
• la fermentazione, la purificazione della proteina, l’analisi qualitativa, la formulazione del prodotto e la conservazione.
Le autorità regolatorie (internazionali e nazionali) richiedono correttamente al produttore di farmaci biotecnologici di eseguire un accurato monitoraggio di tutte le differenti fasi del processo di produzione e purificazione del farmaco, in modo da garantire al massimo le proprietà cliniche e biologiche del prodotto finale.
Per “farmaco biosimilare” si intende, secondo la definizione dell’EMA (European Medicines Agency), un farmaco biotecnologico che è simile, ma non l’esatta copia, di un prodotto biotecnologico registrato, ma non più coperto da brevetto (farmaco “di riferimento” o “originator”) e che è in grado di garantire gli stessi effetti terapeutici e lo stesso grado di tollerabilità del farmaco “di riferimento”.
I farmaci biosimilari sono approvati dalla Commissione Europea (CE) mediante una procedura centralizzata sotto la supervisione dell’EMA. Come tutti i medicinali, anche i farmaci biosimilari sono soggetti a normative e linee guida che stabiliscono i requisiti di qualità, efficacia e sicurezza che tali farmaci devono avere. La qualità si ottiene attraverso rigorosi standard di produzione e severi controlli sulla stessa.
Due aspetti essenziali da valutare in relazione alla qualità sono l’attività e la purezza del prodotto, che non devono differire in maniera significativa dal prodotto di riferimento.
Il processo di sviluppo dei farmaci biosimilari avviene attraverso processi produttivi più recenti, alcuni dei quali innovativi rispetto al medicinale di riferimento, prodotto con tecniche risalenti all’epoca della registrazione di brevetto.
I farmaci biosimilari sono sottoposti ad una attenta valutazione della loro comparabilità con il prodotto di riferimento. La valutazione viene effettuata caso per caso, per ogni prodotto biosimilare. Inoltre, come avviene per tutti gli altri, i biosimilari, una volta approvati, vengono monitorati permanentemente, per garantire una continua sicurezza.
I dati relativi alla sicurezza dei pazienti sono raccolti mediante rigorose attività di farmacovigilanza, che includono misure di routine e monitoraggio specifiche, come illustrato in dettaglio nel Piano di Gestione del Rischio (Risk Management Plan) approvato dall’EMA.
Uno dei settori in cui maggiormente si è sviluppato negli ultimi anni il mercato dei farmaci biosimilari è quello delle epoetine umane ricombinanti.
Esse hanno una sequenza peptidica identica all’EPO umana che, come noto, è una glicoproteina responsabile della stimolazione e della regolazione dell’eritropoiesi. L’EPO umana viene prodotta principalmente nei reni dai fibroblasti peritubulari e si lega a specifici recettori di membrana dei precursori eritroidi nel midollo osseo, promuovendo la loro differenziazione in globuli rossi.
Le EPO ricombinanti vengono prodotte mediante tecnologia di ingegneria genetica in cellule ovariche del criceto cinese (CHO) e sono in grado di svolgere la stessa attività biologica dell’EPO umana. Esiste tuttavia una variabilità nella glicosilazione tra le diverse epoetine; infatti, il corredo enzimatico delle cellule CHO differisce da quello dei fibroblasti renali umani e, inoltre, la diversa glicosilazione dipende anche dal vettore, dalle condizioni ambientali di coltura e dal processo di purificazione. Va altresì ricordato che sia l’EPO umana, che le epoetine ricombinanti sono miscele di isoforme a diverso contenuto di acido sialico.
L’EMA ha emanato una direttiva specifica per quanto riguarda le epoetine ricombinanti; la comparabilità con il prodotto di riferimento deve essere dimostrata in almeno 2 studi:
• randomizzati, a gruppi paralleli
• preferibilmente in doppio cieco
• su pazienti con anemia secondaria ad IRC
• con somministrazione sia e.v., sia s.c.
• con dati di immunogenicità per un periodo minimo di 1 anno.
L’epoetina zeta è un farmaco biotecnologico, prodotto da Norbitec (Germania), che viene ottenuto, analogamente al prodotto originatore (epoetina alfa), mediante tecniche di ingegneria genetica da cellule CHO e ha struttura primaria identica all’EPO umana.
Epoetina zeta possiede una minore quantità di varianti non desiderate dell’acido sialico rispetto al prodotto di riferimento. Un’altra peculiarità di epoetina zeta è l’eccipiente polisorbato 20, che viene utilizzato come agente tensioattivo in luogo del polisorbato 80 e consente di valutare meglio il grado di purezza del prodotto. Inoltre, i prodotti che utilizzano come eccipiente polisorbato 20, a differenza di quelli che usano polisorbato 80, non sono mai stati associati a casi di aplasia pura della serie rossa (PRCA) nel corso del trattamento. Attraverso un programma esteso di caratterizzazione molecolare, è stata dimostrata la similarità di epoetina zeta riguardo alla struttura proteica e alla qualità del prodotto. In accordo con le richieste del processo autorizzativo, è stata condotta una serie di studi preclinici (test in vitro e in vivo), che hanno chiaramente dimostrato la biosimilarità in termini di farmacodinamica, tossicità e antigenicità di epoetina zeta ed epoetina alfa (Figura 1).
In accordo con le richieste dell’EMA, epoetina zeta è stata altresì valutata in studi clinici per dimostrarne l’equivalenza terapeutica ed il profilo di sicurezza rispetto al prodotto di riferimento, cioè l’epoetina alfa.
A tale scopo, epoetina zeta è stata valutata sia in studi di farmacocinetica (dopo somministrazione endovenosa e sottocutanea) che in studi di terapia dell’anemia in corso di insufficienza renale.
FIGURA 1. Epoetina zeta e processo autorizzativo |
Il primo studio di farmacocinetica è stato condotto con l’obiettivo di confrontare i parametri farmacocinetici di epoetina zeta ed epoetina alfa, dopo singola somministrazione di un bolo endovena di 10.000 UI in soggetti sani, e di valutare la sicurezza d’impiego dei due farmaci.
Il disegno del trial, monocentrico, in aperto, randomizzato, crossover, in due fasi, ha previsto un periodo di wash-out della durata di 10 giorni tra la somministrazione dei due farmaci in studio, nonché la determinazione dei livelli di epoetina plasmatica basale e a diversi intervalli dalla somministrazione di ciascuna epoetina ricombinante. I risultati dello studio, relativi a 21 soggetti, hanno evidenziato che i parametri farmacocinetici di epoetina zeta sono comparabili a quelli dell’epoetina alfa (Figura 2); inoltre, entrambe le epoetine sono state ben tollerate.
L’obiettivo di un secondo trial randomizzato, in doppio cieco, crossover, in 3 fasi, è stato di confrontare la biodisponibilità di epoetina zeta somministrata per via sottocutanea (s.c.), con quella dello stesso prodotto somministrato per via endovenosa (e.v.) e quella della epoetina alfa somministrata sempre per via sottocutanea.
FIGURA 2. Curva di concentrazione plasmatica dopo somministrazione e.v. di un bolo di 10.000 UI di epoetina zeta e di epoetina alfa Kirkov V. et al. Alzneimittel Forschung 2008; 58: 215-219 |
I valori della biodisponibilità e dell’emivita di epoetina zeta somministrata per via s.c. sono risultati corrispondenti a quelli riportati per l’epoetina alfa; analogamente, le curve di concentrazione dell’eritropoietina a seguito della somministrazione di una singola iniezione di 10.000 UI s.c. di epoetina zeta e di epoetina alfa sono risultate sovrapponibili. La tollerabilità locale e generale di entrambi i prodotti è risultata buona.
In base ai risultati di questi due studi di farmacocinetica è stato pertanto possibile dimostrare, in accordo alle richieste delle autorità regolatorie, che le proprietà farmacocinetiche di epoetina zeta non differiscono sostanzialmente da quelle del farmaco di riferimento epoetina alfa.
Dopo gli studi di farmacocinetica, sono stati condotti studi clinici di terapia dell’anemia in corso di insufficienza renale, che hanno confermato l’equivalenza terapeutica tra epoetina zeta e prodotto di riferimento. Di seguito vengono illustrati gli studi clinici maggiormente significativi.
Studio di correzione Studio randomizzato, in doppio cieco, multicentrico, di fase III, con l’obiettivo primario di valutare l’equivalenza terapeutica di epoetina zeta ed epoetina alfa nella correzione dei livelli di emoglobina in pazienti affetti da anemia conseguente ad insufficienza renale cronica in stadio avanzato ed in trattamento emodialitico (n=609). Dopo un periodo di run-in di 6 settimane, i pazienti sono stati assegnati in maniera randomizzata al trattamento con epoetina zeta (n=305) o con epoetina alfa (n=304) somministrate e.v. 1-3 volte a settimana per 24 settimane.
I risultati dello studio hanno dimostrato che epoetina zeta è terapeuticamente equivalente all’epoetina alfa, in quanto ha permesso di correggere i livelli di emoglobina in maniera non statisticamente differente rispetto all’epoetina alfa, mostrando inoltre un profilo di tollerabilità comparabile a quello dell’epoetina di riferimento (Figura 3).
Studio di mantenimento Trial randomizzato, in doppio cieco, cross-over, multicentrico, di fase III, in pazienti emodializzati affetti da anemia secondaria ad insufficienza renale, che ha previsto due fasi: una in aperto di run-in e l’altra in doppio cieco. Durante il periodo di run-in, tutti i pazienti hanno ricevuto epoetina alfa e.v. da 1 a 3 volte a settimana per 12- 16 settimane per raggiungere i criteri di randomizzazione nella fase di doppio cieco successiva (livelli di emoglobina compresi tra 10,5 e 12,5 g/dl con una dose costante di epoetina alfa e nessuna variazione intraindividuale di emoglobina superiore a 0,6 g/dl in 4 settimane); i pazienti sono stati quindi assegnati in maniera randomizzata al trattamento con epoetina zeta (n=155) o con epoetina alfa (n=158) somministrate e.v. 1-3 volte a settimana per 12 settimane, dopo le quali 146 pazienti del gruppo trattato con epoetina zeta e 145 pazienti trattati con epoetina alfa sono stati sottoposti al trattamento alternativo per ulteriori 12 settimane. I risultati di questo trial hanno dimostrato che epoetina zeta, somministrata per via e.v. è equivalente dal punto di vista terapeutico all’epoetina alfa nel mantenere costanti i livelli di emoglobina, mostrando un profilo di sicurezza e tollerabilità analogo al farmaco di riferimento (Figura 4).
FIGURA 3. Concentrazione media settimanale di emoglobina durante il periodo di trattamento dello studio (popolazione per-protocol, n=541) Current Medical Research and Opinion® Vol. 24, No. 5, 2008; 1407–1415 |
FIGURA 4. Concentrazione media di emoglobina (Hb) in pazienti con anemia da insufficienza renale cronica emodializzati durante la fase di run-in (trattamento con epoetina alfa) e la fase di trattamento in doppio cieco (pazienti randomizzati al trattamento con epoetina alfa o epoetina zeta) Wizemann et al. Curr Med Res 2008; 24(3): 625-37 |
Studio di follow-up a lungo termine sulla sicurezza d’impiego e tollerabilità Studio in aperto, non controllato, di follow-up, multicentrico, condotto nei pazienti con anemia da insufficienza renale cronica in trattamento emodialitico che avevano completato il periodo di trattamento in doppio cieco dei due precedenti studi di correzione e di mantenimento.
I risultati hanno dimostrato la sicurezza a lungo termine di epoetina zeta e in particolare non è emersa alcuna relazione tra somministrazione di epoetina zeta e comparsa di anticorpi anti-eritropoietina, né è stato osservato alcun fenomeno di resistenza all’epoetina; nello stesso studio è stato inoltre dimostrato che epoetina zeta è in grado di mantenere stabili i livelli di emoglobina entro il range target di 10,5-12,5 g/dl mediante l’impiego di dosaggi che sono rimasti pressoché costanti durante l’intero periodo di follow-up (Figura 5).
Studi osservazionali indipendenti Segnalazioni di conferma dell’efficacia e della tollerabilità di epoetina zeta sono state fornite da diversi studi osservazionali indipendenti. In particolare, l’esperienza personale dell’autore di questa relazione, riferita a 8 pazienti in HD convertiti da darbepoetina alfa (n=5) o epoetina alfa (n=3) a epoetina zeta, ha evidenziato in un periodo di osservazione fino a 18 mesi un costante andamento temporale dei livelli di emoglobina dopo lo switch a epoetina zeta.
Studi sull’impiego s.c. Un primo studio, in aperto, non controllato, multicentrico, di fase III, in cui sono stati arruolati 216 pazienti, ha evidenziato che epoetina zeta somministrata per via sottocutanea risulta efficace e ben tollerata nel trattamento dell’anemia in pazienti affetti da neoplasia in trattamento chemioterapico e a rischio di trasfusione.
Inoltre, è attualmente in corso uno studio osservazionale internazionale multicentrico, di ampie dimensioni (n=6700 pazienti) che ha come obiettivo principale quello di stimare l’incidenza di PRCA, anticorpi neutralizzanti, perdita di efficacia ed eventi tromboembolici, per una durata del follow-up di 3 anni, nei pazienti trattati con epoetina zeta s.c.; questo studio, denominato PASCO II, costituisce uno dei requisiti dell’approvazione all’uso s.c. da parte dell’EMA.
FIGURA 5. Concentrazione di emoglobina (Hb) e dosaggio di epoetina zeta nel corso dello studio di follow-up in aperto (n=745) Baldamus et al. Adv Ther. 2008; 25(11) |
Studi post-marketing I dati post-marketing più recenti disponibili (aggiornati al 31/12/2011) indicano che il numero di soggetti esposti all’epoetina zeta supera gli 86.000 pazienti e che non sono stati riscontrati particolari problemi di sicurezza del prodotto, né è stato riportato sinora alcun caso di sviluppo di anticorpi neutralizzanti o di PRCA.
In conclusione, i dati preclinici e clinici forniscono delle indicazioni assolutamente confortanti in merito all’equivalenza terapeutica rispetto al prodotto di riferimento e alla sicurezza di impiego di epoetina zeta.
Infine un aspetto importante da evidenziare, e che viene preso in considerazione anche durante gli studi pre-marketing, riguarda la qualità. Per esempio, in uno studio di Brinks et al., che ha confrontato la qualità di varie eritropoietine (EPO), inclusi due biosimilari, per quanto riguarda contenuto, aggregazione, profilo di isoforme e potenza, Retacrit (epoetina zeta) è risultato una miscela di 6 isoforme come il prodotto di riferimento.
Una tematica da non sottovalutare, e che probabilmente avrà un sempre maggiore interesse nel prossimo futuro, è rappresentata dagli aspetti economico-finanziari del trattamento con biosimilari.
Va ricordato che i farmaci biotecnologici, in generale, hanno un costo elevato; basti pensare che, se si considerano la spesa e i consumi 2010 per farmaci erogati dalle strutture pubbliche, risulta che le eritropoietine (ESA) hanno comportato una spesa pubblica di 392 milioni di Euro (2% della spesa farmaceutica a carico del SSN); nello stesso anno, il 36% della spesa legata alla categoria terapeutica “Sangue e organi ematopoietici” è stato dovuto alla prescrizione delle ESA.
Ci si attende che i biosimilari potrebbero esercitare un significativo impatto positivo in termini di risparmio della spesa sanitaria, con due meccanismi:
• Risparmio diretto per il minor costo di acquisto
• Effetto competitivo sul costo degli originatori. Va detto che al momento non sono disponibili studi di farmacoeconomia che dimostrino concretamente vantaggi in termini di riduzione delle spese sanitarie con i biosimilari rispetto alle epoetine “originators”.
FIGURA 6. Costi: esperienza personale. I numeri all'interno degli istogrammi riportano la percentuale di utilizzo di epoetina alfa biosimilare rispetto al numero totale dei pazienti trattati con ESA 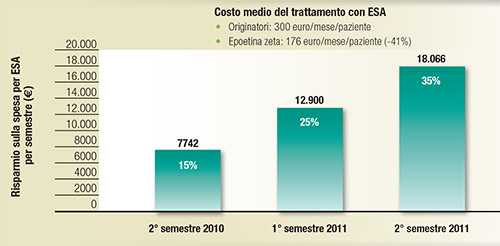 |
Un dato interessante proviene comunque dall’esperienza personale dell’autore di questa relazione, secondo cui, nel periodo 2010-2011, il costo medio del trattamento con epoetine “originators” è stato di 300 euro/mese/paziente e di 176 euro/mese/paziente con epoetina zeta, con un risparmio quindi del 41% con epoetina zeta (Figura 6).
Sulla base dell’esperienza maturata in Germania, dove nel primo anno di introduzione dei biosimilari il risparmio è stato di 60 milioni di euro (pari al 17,3% della spesa per epoetine), si stima che anche in Italia potrà aversi nei prossimi anni, con una maggiore diffusione dei biosimilari, una significativa e progressiva riduzione della spesa globale legata all’utilizzo di eritropoietine (Figura 7).
Va comunque tenuto presente che la spesa sanitaria nella realtà pratica viene condizionata da diverse variabili, tra cui:
• la sovrapponibilità di efficacia: da questo punto di vista epoetina zeta ha ampiamente dimostrato la propria equivalenza terapeutica con l’epoetina di riferimento (alfa);
• il comportamento prescrittivo, che viene influenzato dalle differenti direttive dei Sistemi Sanitari nei vari Paesi europei (ad esempio, nel Regno Unito le prescrizioni devono essere in accordo con Linee Guida a carattere nazionale, mentre in Italia vi è una maggiore diversificazione nelle varie Regioni in relazione a delibere regionali).
| FIGURA 7. Stima del contributo dei biosimilari al risparmio per le eritropoietine in Italia Jommi C. Biosimilari 2010; 1:19-30 |
CONCLUSIONI
Lo sviluppo di un biosimilare nell’Unione Europea è un processo rigoroso dal punto di vista scientifico, con barriere regolatorie molto severe. Ciò, associato ai processi di farmacovigilanza attiva, assicura che solo prodotti di alta qualità possano entrare nei prontuari terapeutici europei, incluso quello italiano.
• L’epoetina zeta, biosimilare dell’epoetina alfa, ha superato tutte le severe “barriere” del processo autorizzativo europeo ed è oggetto di stretto monitoraggio post-marketing allo scopo di confermare attivamente la sua efficacia e sicurezza nella pratica clinica.
• I risultati degli studi clinici hanno ampiamente confermato che l’epoetina zeta è equivalente dal punto di vista terapeutico all’epoetina alfa, il prodotto di riferimento.
• I dati post-marketing, relativi ad oltre 86.000 pazienti trattati dal 2008 con l’epoetina zeta, confermano la sicurezza di impiego del biosimilare, indicando che non sono stati riscontrati particolari problemi di sicurezza, né è stato riportato sinora alcun caso di sviluppo di anticorpi neutralizzanti o di PRCA.
• Non bisogna dimenticare che l’uso dei biosimilari, sotto lo stretto controllo delle Autorità regolatorie, ha una forte valenza etica, perchè i risparmi ottenibili potranno garantire ai pazienti la disponibilità di farmaci innovativi anche se costosi.
• Pertanto, si può concludere con l’affermazione che con l’utilizzo dei biosimilari gli interessi di pazienti, enti regolatori e SSN, i tre componenti del “triangolo di portatori di interesse”, tenderanno sempre più a convergere.
|