
La malattia di Gaucher: la meno “rara” delle oltre 50 malattie da accumulo lisosomiale
La malattia di Gaucher, che prende il nome da Philippe Gaucher, un dermatologo che nel 1882 descrisse il caso di una donna di 32 anni con una splenomegalia massiva e cellule insolitamente voluminose nella milza, è una malattia ereditaria autosomica recessiva da accumulo lisosomiale (1).
Oltre ad essere la prima che è stata descritta tra le oltre 50 malattie da accumulo lisosomiale, la malattia di Gaucher, con una frequenza di 1 caso su 40000 persone, è la più comune di questo gruppo di malattie rare (1,2).
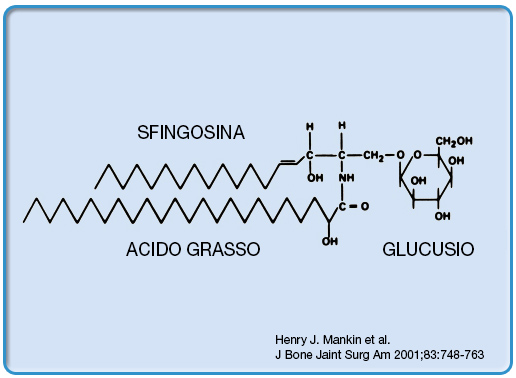
La causa è la carenza dell’enzima lisosomiale β-glucocerebrosidasi, che comporta l’accumulo del substrato, il glucocerebroside glucosilceramide (Figura 1), un importante componente di cellule tessutali e membrane di organuli cellulari, nei lisosomi dei macrofagi.
Figura 1. La glucosilceramide è costituita da sfingosina e un acido grasso |
|---|
 |
I macrofagi ripieni di lipidi (le cellule schiumose di Gaucher) si accumulano in organi che contengono un numero significativo di cellule della linea cellulare macrofagica, quali milza, fegato, osso corticale e midollo osseo, linfonodi e polmoni e, pertanto, i segni della malattia sono correlati alla splenomegalia con ipersplenismo, all’epatomegalia e all’interessamento osseo e, occasionalmente, alla disfunzione polmonare (3).
L’accumulo di glucosilceramide nei lisosomi inoltre determina l’attivazione dei macrofagi, evidenziata dagli elevati livelli IL-1, IL-6, IL-1 e TNF-α presenti nei pazienti. Tali citochine sono coinvolte in numerosi processi fisiologici e potrebbero essere responsabili di conseguenze quali ad es. aumentata attività osteoclastica con osteopenia, osteoporosi e fratture (4).
A livello genetico, la malattia di Gaucher è causata dalla mutazione di due alleli del gene GBA1 che codifica per la glucocerebrosidasi. I quattro alleli più comuni complessivamente sono responsabili di più del 96% delle mutazioni nei pazienti ebrei Ashkenazi, mentre si ritrovano in meno del 75% dei pazienti di altri Paesi occidentali. La frequenza di ognuna delle 320 mutazioni responsabili della malattia, inoltre, varia tra popolazioni differenti. Nella maggior parte dei casi, le mutazioni sono di tipo missense e determinano la riduzione dell’attività catalitica e/o della stabilità enzimatica (5).
I fenotipi, per essendo come per molte malattie genetiche un continuum di gradi di interessamento, possono essere raggruppati in due varianti, la non neuropatica (tipo 1) e la neuropatica (tipo 2 e tipo 3). All’interno di tali categorie esiste una notevole eterogeneità fenotipica, con forme più gravi e forme attenuate, ed espressione e gravità possono variare perfino tra fratelli (sibling) con lo stesso genotipo (2, 5). La malattia di tipo I, che costituisce il 95% dei casi, presenta manifestazioni variabili limitate agli organi viscerali e può manifestarsi a qualunque età. La malattia di tipo II e la malattia di tipo III si distinguono per età di esordio, rapidità e gravità della malattia progressiva a carico del sistema nervoso (3).
Il genotipo del paziente può fornire informazioni sull’evoluzione della malattia, sui risultati attesi dal trattamento e rappresentare una base per il counselling genetico (Figura 2) (1).
Figura 2. Il genotipo del paziente è importante sia ai fini prognostici sia come base per il counselling genetico e la pianificazione familiare. |
|---|
 |
| Bibliografia |
|---|
1. Grabowski GA. Gaucher disease and other storage disorders. Hematology Am Soc Hematol Educ Program. 2012;2012:13-18
2. Martins AM, Ribeiro Valadares E, Porta G et al. Recommendations on Diagnosis, Treatment, and Monitoring for Gaucher Disease. The Journal of Pediatrics 2009; 155 (4, Suppl.): S10–8
3. Burrow TA, Grabowski GA. Velaglucerase alfa in the treatment of Gaucher disease type 1. Clin Investig (Lond) 2011; 1(2): 285–293
4. Harmanci O, Bayraktar Y. Gaucher disease: New developments in treatment and etiology. World J Gastroenterol 2008; 14(25): 3968-3973
5. Burrow TA, Barnes S, Grabowski GA. Prevalence and management of Gaucher disease. Pediatric Health, Medicine and Therapeutics 2011; 2: 59–73
N. 2/2015 - MedTOPICS - Periodico Quindicinale |